In June 2026, a 23-year-old man named Daniel Cressy rang a ceremonial bell at Manning Family Children’s Hospital in New Orleans, surrounded by his medical team, family, and the mayor of New Orleans. He had just become the first person in the US Gulf Coast region to be declared functionally cured of sickle cell disease, a condition he had lived with since infancy. For most of his life, sickle cell disease stood between him and his childhood dream of flying commercial jets. The Federal Aviation Administration would not license a pilot with the condition, since the altitudes at which commercial aircraft fly can trigger dangerous complications in people with sickled red blood cells. Today, that dream is finally within reach.
Cressy’s story is spreading fast, and it deserves to. It’s a genuine celebration, a young man reclaiming a future that a lifelong disease once put out of reach, made possible by one of the most remarkable applications of gene-editing science to date. And the science behind it is worth understanding, because it’s not a one-off miracle. It’s the product of decades of careful research into how our bodies produce blood, brought to life through a sickle cell gene therapy called Casgevy, now helping patient after patient reach the same milestone Cressy just celebrated.
This article walks through what this sickle cell cure actually is, how the underlying CRISPR gene therapy works, what the clinical evidence shows, and what it means for the future of sickle cell disease treatment.
What sickle cell disease actually does to the body
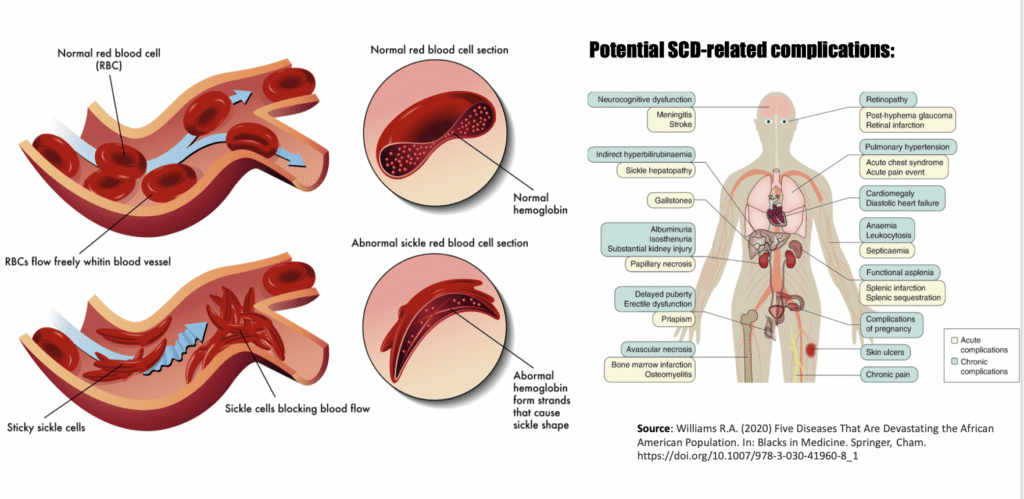
Sickle cell disease is a genetic blood disorder caused by a single mutation in the gene that codes for beta-globin, one of the two protein chains that make up hemoglobin, the molecule in red blood cells responsible for carrying oxygen. That one mutation produces an abnormal form of hemoglobin called hemoglobin S, or HbS.
Under normal conditions, red blood cells are flexible, disc-shaped, and squeeze easily through even the narrowest blood vessels. But when hemoglobin S loses its oxygen, it clumps together inside the red blood cell, distorting the cell into a rigid, crescent, or “sickle,” shape. These misshapen cells are prone to getting stuck in small blood vessels, blocking blood flow and starving tissue of oxygen. The result is what’s known as a vaso-occlusive crisis, or VOC, a sudden, often severe pain episode that can require emergency hospitalization. Over years, repeated VOCs damage organs, and the disease overall shortens life expectancy.
Sickle cell disease predominantly affects people of African descent, along with communities across the Mediterranean, Middle East, and South Asia, regions where the sickle cell trait historically offered some protection against malaria. In the United States, it affects roughly 100,000 people, the overwhelming majority of them Black. For much of modern medical history, treatment options were limited to managing symptoms: pain medication, blood transfusions, and a drug called hydroxyurea that reduces crisis frequency but doesn’t address the underlying cause.
How Casgevy Works: The Science Behind the Sickle Cell Gene Therapy
The treatment Cressy received is called Casgevy, generic name exagamglogene autotemcel (often shortened to exa-cel). It’s a one-time cell therapy built on CRISPR-Cas9, the gene-editing technology whose inventors won the Nobel Prize in Chemistry in 2020. Casgevy was the first CRISPR-based therapy ever approved for human use, cleared by the FDA for sickle cell disease in December 2023.
Here’s the elegant part of how it works. Rather than trying to fix the faulty beta-globin gene directly, which is technically very difficult to do precisely, researchers took a different route: reactivating a gene that the body normally switches off after birth.
Every human starts life producing a different form of hemoglobin, fetal hemoglobin (HbF), during gestation. Shortly after birth, a gene called BCL11A switches on and suppresses fetal hemoglobin production, redirecting the body toward adult hemoglobin production instead. In people with sickle cell disease, that adult hemoglobin is the defective HbS.
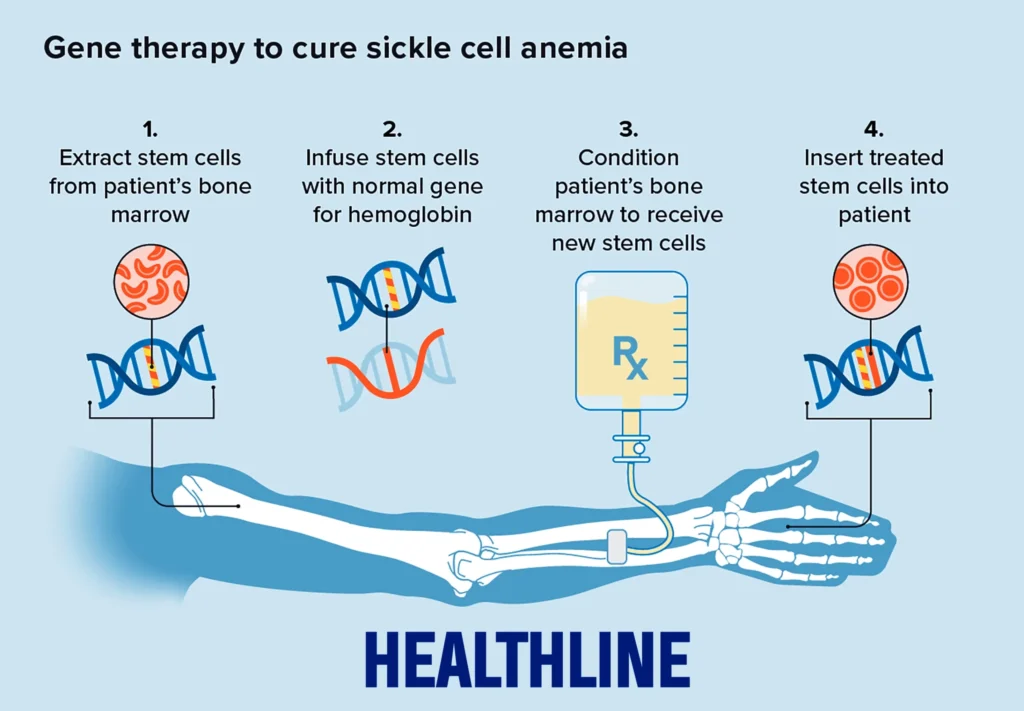
Casgevy works by extracting a patient’s own blood-forming stem cells, then using CRISPR-Cas9 to make a precise cut at a specific regulatory region of the BCL11A gene, called the erythroid-specific enhancer. This edit disrupts a binding site that the protein GATA1 needs to activate BCL11A. With BCL11A suppressed, the edited stem cells resume producing fetal hemoglobin, alongside residual defective hemoglobin. Because fetal hemoglobin doesn’t sickle, red blood cells stay flexible and functional, effectively bypassing the disease’s underlying mechanism rather than repairing it directly.
What patients actually go through
The process is neither quick nor simple. Cressy’s own path took roughly two years from decision to bell-ringing. It generally unfolds in stages:
Cell collection. Patients first undergo regular blood transfusions in preparation, then their own blood-forming stem cells are mobilized out of the bone marrow and into the bloodstream using a drug called plerixafor, and collected through a process called apheresis, sometimes over multiple sessions.
Gene editing. The collected cells are shipped to a specialized manufacturing lab, in Cressy’s case, to Scotland, where the CRISPR-Cas9 editing takes place outside the body. This is what makes the therapy “ex vivo”: the actual gene editing never happens inside the patient.
Conditioning. Before the edited cells can be returned, the patient’s bone marrow needs to be cleared to make room for them. This involves several days of high-dose chemotherapy (busulfan), which eliminates the existing, disease-producing stem cells. This step carries real risks and side effects, including infertility risk and a period of extreme vulnerability to infection, since the immune system is essentially wiped out temporarily.
Infusion and engraftment. The genetically modified cells are infused back into the patient intravenously. Over the following weeks, they need to “engraft,” settling into the bone marrow and beginning to produce blood cells. Patients typically spend around a month in inpatient recovery while doctors monitor blood counts to confirm the new cells have taken hold.
Because the edited cells are the patient’s own, there’s no risk of transplant rejection, a key difference from older sickle cell treatments that relied on bone marrow donated by a matched donor, which came with graft-versus-host disease risk and depended on finding a compatible donor in the first place.
The Clinical Trial Results Behind the Sickle Cell Breakthrough
Casgevy’s approval rested on a Phase 3 clinical trial (known as CLIMB-121) involving patients aged 12 to 35 who had experienced at least two severe vaso-occlusive crises per year for two years prior to treatment, a population with genuinely severe, treatment-resistant disease. The final results, published in the New England Journal of Medicine in April 2024, found that of 31 evaluable patients, 29 (roughly 93.5%) experienced no severe vaso-occlusive crisis for at least 12 consecutive months following treatment. Of those evaluated for hospitalization, 100% avoided any sickle-cell-related hospital stay over the same period. A separate NEJM analysis of off-target editing effects, examining thousands of candidate sites across the genome, found no evidence of unintended edits elsewhere, an important safety signal for a technology this new.
Follow-up studies since approval have extended these findings to younger patients, including a cohort aged 5 to 11, presented at the European Hematology Association Congress and published simultaneously in NEJM, showing consistent efficacy and safety in that age group as well.
Why “functional cure” is the accurate phrase, not just a softer one
It’s worth being precise about the terminology, because it matters. Casgevy doesn’t repair the mutated gene itself, and it doesn’t remove sickle hemoglobin from a patient’s body entirely; edited cells still carry the underlying beta-globin mutation. What changes is that those cells now also produce enough fetal hemoglobin to prevent the sickling process that causes disease symptoms. Patients are described as “functionally cured” because, by the clinical measures that matter most, freedom from pain crises, hospitalizations, and organ damage, their disease effectively stops behaving like sickle cell disease. Whether that protection holds for decades is still being studied, since the therapy has only been in real-world use since late 2023.
The Road Ahead for Sickle Cell Disease Treatment
This gene therapy breakthrough is genuinely exciting, and the momentum behind it is real: more hospitals are gaining approval to offer it, more patients are completing treatment, and researchers are already extending the data to younger age groups. A few things are worth knowing about where things stand today, for the roughly 20 million people worldwide living with sickle cell disease who are watching this progress closely.
Cost and access. Gene therapies like Casgevy carry list prices in the millions of dollars per treatment, which puts them out of reach for the vast majority of patients globally, including in regions of sub-Saharan Africa and South Asia where the disease burden is highest.
Eligibility. The approved treatment is currently limited to patients aged 12 and older with frequent, severe vaso-occlusive crises, not the full population of people living with sickle cell disease.
The conditioning process itself carries risk. The chemotherapy required to clear existing bone marrow is intensive, and recovery is not without complications.
Manufacturing complexity. Because each dose is manufactured individually from a single patient’s own cells, shipped internationally in some cases, and takes months to prepare, scaling this approach to treat millions of patients presents a very different challenge than manufacturing a conventional drug.
The Bigger Picture: A Milestone Worth Celebrating
Daniel Cressy’s journey, from a childhood diagnosis that seemed to close off his dream of flying, to ringing a bell as a functionally cured man with that dream back on the table, is exactly the kind of story that makes the promise of gene therapy feel real and personal. It’s the human face of a scientific achievement years in the making: reactivating a fetal gene switched off at birth to correct a disease that has affected millions of lives for generations. Every patient who completes this sickle cell cure and rings that bell adds to a growing body of evidence that this approach works, and every regional first, like Cressy’s, means the therapy is reaching further, faster. That’s genuinely worth celebrating, and it’s a strong sign of where sickle cell disease treatment is headed next.
References
Vargas, R.A. (2026, June 24). Louisiana man becomes first in region functionally cured of sickle cell disease. The Guardian. https://www.theguardian.com
Frangoul, H., et al. (2024). Exagamglogene Autotemcel for Severe Sickle Cell Disease. New England Journal of Medicine. DOI: 10.1056/NEJMoa2309676
U.S. Food and Drug Administration. (2023, December 8). FDA approval letter for exagamglogene autotemcel (Casgevy). https://www.fda.gov/media/175179/download?attachment
Children’s Hospital of Philadelphia. (2025). Researchers Publish Final Results of Key Clinical Trial for Gene Therapy for Sickle Cell Disease. https://www.chop.edu
Boston Children’s Hospital. CASGEVY (Exagamglogene Autotemcel) — Clinical Overview. https://www.childrenshospital.org/conditions-treatments/casgevy
Vertex Pharmaceuticals. (2026). Vertex Presents New Data on CASGEVY, Including First European Presentation of Data in Children Ages 5–11. https://news.vrtx.com
NCBI Bookshelf. Clinical Review — Exagamglogene Autotemcel (Casgevy). https://www.ncbi.nlm.nih.gov/books/NBK614940/
Specificity of CRISPR-Cas9 Editing in Exagamglogene Autotemcel. (2024). New England Journal of Medicine. DOI: 10.1056/NEJMc2313119